GERM CELL TUMORS
GCTs account for approximately one-third of all pineal tumors and are histologically
identical to gonadal GCTs with a predominance in men and younger age groups. Extragonadal
GCTs (including pineal region) have a poorer prognosis than gonadal GCTs. GCTs range along
a spectrum from benign (as in teratomas, dermoids, epidermoids, and lipomas) to highly
malignant (as in choriocarcinomas, embryonal cell carcinomas, teratocarcinomas, and
endodermal sinus tumors).
Germinomas are tumors of an intermediate degree of malignancy arising from primordial germ
cells that can occur in the gonads or in midline sites in the nervous system (pineal and
suprasellar region) or body (mediastinum and sacrococcygeal region). Although
histologically identical in all sites of origin, by conventional nomenclature germinomas
in the testes are called seminomas, those in the ovaries are dysgerminomas, and those in
the CNS are germinomas (previously called atypical teratomas).
Unlike suprasellar GCTs, which show no sexual predisposition, GCTs of the pineal region
occur predominantly in males. Germinomas are most common in boys in the first or second
decade and have a propensity to seed the cerebrospinal fluid (CSF) pathways. Despite their
malignant characteristics, germinomas are exquisitely sensitive to radiotherapy and
chemotherapy; they can be cured in many patients. The presence of beta human chorionic
gonadotropin (b-hCG) in a germinoma implies a slightly worse prognosis.Embryonal cell
carcinomas, choriocarcinomas, and endodermal sinus tumors are rare but highly malignant
GCTs that may metastasize to the CSF. Choriocarcinoma contains cyto- and
syncytiotrophoblastic cells that produce b-hCG. Endodermal sinus tumors contain yolk sac
elements that produce alpha-fetoprotein (AFP). High levels of b-hCG or AFP in the CSF or
serum indicate the presence of malignant germ cell elements, and histologic confirmation
is not necessary for treatment with radiation, chemotherapy, or both . GCTs in general are
difficult to classify because 25% are of a mixed type, containing both malignant and
benign elements or several different malignant elements. Extensive specimen sampling is
necessary for accurate histologic determination. High AFP or b-hCG levels in the CSF
trigger an extensive search for malignant germ cell elements even if the pathologic
specimens suggest otherwise. Benign GCTs such as teratomas, dermoids, and epidermoids are
generally curable with surgery alone. Teratomas are composed of tissues from three germ
cell lines (endo-, ecto-, and mesoderm). Immature teratomas are a variant of this tumor
that may grow quickly and behave in a malignant fashion, including CSF seeding.
PINEAL CELL TUMORS
Pineal cell tumors arise from pineocytes and range from histologically primitive
pineoblastomas to well-differentiated pineocytomas. Attempts to correlate prognosis and
survival with either variant have been inconclusive because both may behave in a malignant
fashion, with recurring at the primary site and spreading through the CSF. Pineal cell
tumors occur in children and young adults before age 40 with no sex predominance. They are
occasionally found concurrently with retinoblastomas and may contain mixed cell types. The
pineoblastomas are considered a variant of primitive neuroectodermal tumors. Pineal cell
tumors are radiosensitive, but experience with chemotherapy is limited.
GLIOMAS
Gliomas, like GCTs, account for one-third of pineal tumors. Most are invasive and have a
prognosis comparable with astrocytomas of the brainstem. About one-third of gliomas are
low grade, cystic, and surgically curable. Anaplastic astrocytomas and glioblastomas are
less common. Oligodendrogliomas and ependymomas may also occur. Treatment of these tumors
is identical to the treatment of gliomas in other areas of the CNS.
MENINGIOMAS
Meningiomas can arise from the velum interpositum or from the tentorial edge with a higher
incidence in middle age and the elderly. They are amenable to surgical resection.
METASTASIS AND OTHER MISCELLANEOUS TUMORS
The pineal gland does not have a blood–brain barrier and, like the pituitary gland,
may be underrecognized as a possible site for CNS metastasis of systemic tumors.
Miscellaneous tumors include sarcoma, hemangioblastoma, choroid plexus papilloma,
lymphoma, and chemodectoma.
PINEAL CYSTS
Benign cysts of the pineal gland are often found incidentally on radiographic studies, and
it is important to distinguish them from cystic tumors. They are normal variants of the
pineal gland and consist of a cystic structure surrounded by normal pineal parenchymal
tissue. Radiographically they are up to 2 cm in diameter and often have some degree of
peripheral enhancement that may be a compressed normal pineal gland. Pineal cysts may be
found in 4% of all magnetic resonance images. These cysts are static anatomic variants and
need no treatment unless they become symptomatic. In one series of 53 pineal cysts, fewer
than 10% developed hydrocephalus requiring surgical intervention.
SYMPTOMS
Pineal region tumors can become symptomatic by three mechanisms: increased intracranial
pressure from hydrocephalus, direct brainstem and cerebellar compression, or endocrine
dysfunction. Headache, associated with hydrocephalus, is the most common symptom at onset
and is caused by obstruction of third ventricle outflow at the aqueduct of Sylvius. More
advanced hydrocephalus can result in papilledema, gait disorder, nausea, vomiting,
lethargy, and memory disturbance. Direct midbrain compression can cause disorders of
ocular movements such as Parinaud syndrome (paralysis of upgaze, convergence or
retraction, nystagmus, and light-near pupillary dissociation) or the Sylvian aqueduct
syndrome (paralysis of downgaze or horizontal gaze superimposed upon a Parinaud syndrome).
Either lid retraction (Collier sign) or ptosis may follow dorsal midbrain compression or
infiltration. Fourth nerve palsies with diplopia and head tilt may be seen. Reversibility
is a clue to pathogenesis; eye signs due to hydrocephalus recede promptly after
ventricular shunting. Ataxia and dysmetria can result from direct cerebellar compression.
Endocrine dysfunction is rare, usually arising from secondary effects of hydrocephalus
or tumor spread to the hypothalamic region. Diabetes insipidus occurs in less than 5% of
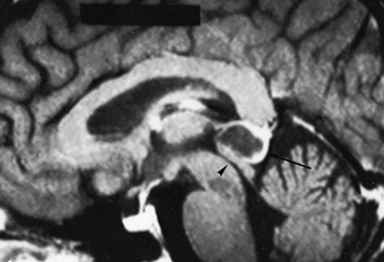
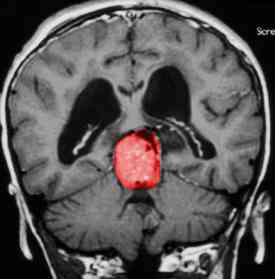
pineal tumors, usually with a germinoma (Fig. 57.2). The symptoms may occur early, before
any radiographic documentation of hypothalamic seeding. Although precocious puberty has
been linked historically with pineal masses, documented cases are rare. Precocious puberty
is actually precocious pseudopuberty because the hypothalamic-gonadal axis is not mature.
It occurs strictly in boys with choriocarcinomas or germinomas with syncytiotrophoblastic
cells and ectopic secretion of b-hCG. In boys, the luteinizing hormonelike effects of
b-hCG can stimulate Leydig cells to produce androgens that induce development of secondary
sexual characteristics and pseudopuberty. This phenomenon does not occur in girls with
pineal region tumors because GCTs are rare in females; also, and more important, both
luteinizing hormone and follicle-stimulating hormone are necessary to trigger ovarian
estrogen production.
DIAGNOSIS
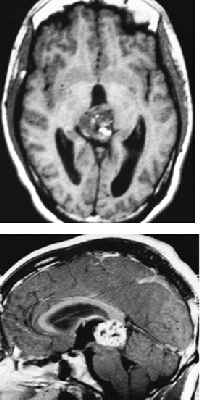
Magnetic resonance imaging (MRI) is the principal diagnostic test for pineal region
tumors. MRI with gadolinium enhancement is mandatory for all pineal tumors to determine
the presence of hydrocephalus and to evaluate tumor size, vascularity, and homogeneity. In
particular, sagittal MRI reveals the relationship of the tumor to surrounding structures
as well as possible ventricular seeding. Computed tomography is complementary but does not
provide as much information as MRI. Angiography is not performed unless a vascular anomaly
is suspected. Measurement of AFP and b-hCG in serum and CSF is routine in the preoperative
workup. If b-hCG or AFP levels are elevated, malignant germ cell elements are present even
if histologic examination gives a benign impression, because a small island of these cells
in a large tumor may be overlooked. Despite improved imaging and CSF markers, a definite
histologic diagnosis cannot be made without pathologic examination of tumor tissue.
SURGERY
Because of the wide variety of pineal region tumor subtypes, a histologic diagnosis is
mandatory for optimal patient management. The pineal region may be approached surgically
from one of several variations, above or below the tentorium. Nearly one-third of pineal
tumors are benign and curable with surgery alone. With malignant tumors, aggressive tumor
resection provides the best opportunity for accurate histologic diagnosis and may increase
the effectiveness of adjuvant radiotherapy or chemotherapy. The overall operative
mortality is about 4%, with an additional 3% permanent major morbidity. The most serious
complication of surgery is hemorrhage into a partially resected malignant tumor. The most
common postoperative complications are ocular palsies, altered mental status, and ataxia;
all are usually transient. For patients with obviously disseminated tumor or those with
medical problems that pose excessive surgical risks, stereotactic biopsy is a reasonable
alternative for obtaining diagnostic tissue. Although gaining in popularity, stereotactic
biopsy is not performed routinely for these reasons: increased sampling error through
insufficient tissue analysis, increased risk of hemorrhage from adjacent deep venous
system and highly vascular pineal tumors, and the better prognosis that follows aggressive
resection.
POSTOPERATIVE STAGING
All patients with pineal cell tumors, malignant GCTs, and ependymomas are thoroughly
evaluated for CSF seeding even though this is a rare occurrence . High-resolution MRI is
more sensitive than computed tomography myelography. CSF cytology is not reliable in
predicting seeding.
RADIATION THERAPY
Radiation therapy consists of 4,000 cGy to the whole brain with an additional 1,500 cGy to
the pineal region; it is recommended for all patients with malignant pineal region tumors.
Spinal radiation is not recommended unless there is radiographic documentation of spinal
seeding.
Radiosurgery for pineal tumors is promising, but experience
is limited. It could be most useful for malignant pineal cell tumors and other small
malignant tumors (less than 3 cm in diameter), particularly if combined with fractionated
radiation. It may also be helpful for tumors that recur after radiation therapy.
Germinomas historically have had an excellent response to external fractionated
radiotherapy, and radiosurgery seems unlikely to improve upon those results.
CHEMOTHERAPY
Chemotherapy has been of most benefit with nongerminomatous malignant GCTs. The most
commonly used regimens are combinations of cisplatin, vinblastine, and bleomycin or
cisplatin and VP-16 (etoposide), which are usually given before radiation therapy. The
results of chemotherapy alone may be comparable with those of radiation therapy for pure
germinomas. There has been a trend to reduce radiation dosage because of long-term
neurotoxicity by combining it with chemotherapy.
LONG-TERM OUTCOME
Generally, benign pineal tumors are curable with surgery alone. Among malignant tumors,
the prognosis depends on the tumor histology. Germinomas have a 75% to 80% 5-year survival
with combined surgery and radiotherapy. Patients with a nongerminomatous malignant GCT
rarely survive beyond 2 years, but this may improve with better chemotherapy. About
one-third of astrocytomas are cystic tumors and are cured by surgery alone. Solid
astrocytomas behave clinically like other brainstem gliomas and have a 67% 5-year survival
rate. Radiation therapy is usually recommended for these tumors, but any effect on
survival is difficult to evaluate. Among pineal cell tumors, a few are discrete,
histologically benign, and completely resectable. Most malignant pineal cell tumors,
however, are not resectable and have a 55% 5-year survival rate with surgery and
radiation.
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}