NEUROFIBROMATOSIS
Neurofibromatosis, an inherited disease transmitted as an autosomal dominant trait, is
noted for its heterogeneity of clinical expression. It may involve the peripheral and
central nervous systems as well as skin and bone, and the endocrine, gastrointestinal, and
vascular systems. Although von Recklinghausen is credited for the initial clinical
description of the disease, it had been described earlier.
Two distinct forms of neurofibromatosis have been recognized: type 1, the most common
type, is widely known as von Recklinghausen disease and is characterized by multiple
hyperpigmented macules (café au lait spots) and neurofibromas; type 2 is characterized by
eighth-nerve tumors (schwannomas), but other intracranial and intraspinal tumors are
common.
The diagnosis of type 1 neurofibromatosis can be made in patients with two or more of the
following clinical findings: six or more café au lait spots with a maximum diameter of
more than 5 mm during prepuberty and more than 15 mm in postpubertal patients; two or more
neurofibromas of any type or one plexiform neuroma; axillary or inguinal freckling; optic
glioma; two or more Lisch nodules; typical bony lesions; and a first-degree relative with
the disorder diagnosed by these criteria. The diagnosis of type 2 neurofibromatosis can be
made if the patient has bilateral eighth-nerve masses as demonstrated by appropriate
imaging techniques. The diagnosis can also be made if the patient has a first-degree
relative with type 2 neurofibromatosis and either a unilateral eighth-nerve mass lesion or
two of the following findings: neurofibroma, meningioma, glioma, schwannoma, or juvenile
posterior subcapsular lenticular opacity.
The skin changes associated with neurofibromatosis are characterized by focal or diffuse
lesions, or both, that are usually present before the appearance of any neurological
abnormality. They include café au lait spots, fibroma molluscum, patchy or diffuse areas
of hyperpigmentation, hypopigmented spots, and angiomas.
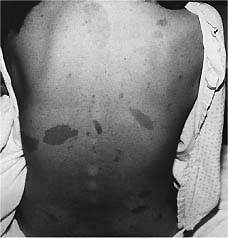
Café au lait spots are usually present at birth. The number of spots and the degree of
pigmentation tend to increase during the first year of life, but after that time the
number of spots remains relatively stable. These spots can appear anywhere on the body
except probably the scalp, palms, and soles. They are typically flat, with discrete
borders, and vary in size from millimeters to centimeters.Crowe and associates observed
that patients with six or more café au lait spots with a diameter of 1.5 cm have a
presumptive diagnosis of neurofibromatosis; Crowe later reported that axillary freckling
was also an important feature of the disease.The presence of café au lait spots does not
necessarily establish the diagnosis of neurofibromatosis, however, as at least 10 percent
of the population have one or more hyperpigmented cutaneous macules.
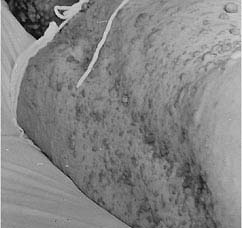
Fibroma molluscum,
a prominent skin lesion found in the dermis or adjacent to it, consists of discrete, soft
or firm papules ranging in size from a few millimeters to several centimeters. They are
flat, sessile, or pedunculated and can be readily impressed into the subjacent skin.
Hypopigmented spots, similar to those found in tuberous sclerosis, are sometimes present,
as are discrete areas of skin hypoplasia and angiomas.
Neurofibromas can occur anywhere from the dorsal root ganglion to the terminal peripheral
nerve branches and can affect any organ system. They vary in size and are more often found
on the trunk than on the limbs Plexiform neuromas are made up of interwoven elements of
tumor and connective tissue that infiltrate normal tissue. They can be superficial,
involving skin and subcutaneous tissues, or deep, affecting visceral and adjacent tissues.
Diffuse areas of skin hyperpigmentation may overlie the plexiform neuroma. The incidence
of sarcomatous transformation of neurofibromas is generally accepted as 2 to 7 percent.
Intracranial tumors, primarily astrocytomas, occur in neurofibromatosis and involve the
cerebrum or cerebellum. There is an increased frequency of optic nerve gliomas, and other
cranial nerves can be affected by neurofibromas or schwannomas. Bilateral acoustic
neuromas are a characteristic feature of type 2 neurofibromatosis. Although meningiomas,
both solitary and multiple, are found with increased frequency in the major type of
neurofibromatosis (type 1), they are more typically observed in the type 2 disorder.
Medulloblastomas, ependymomas, and hamartomas also occur more frequently in patients with
neurofibromatosis than in the general population.
Intraspinal tumors are single or multiple, intradural or extradural. They can be
accompanied by spinal anomalies such as syringomyelia. Some intraspinal tumors assume a
dumbbell shape, extending through the intervertebral foramen. They are sometimes
associated with enlargement of that foramen or defect of the contiguous bone.
Lisch nodules, a typical finding in neurofibromatosis, are melanocytic hamartomas found in
the iris. Age-dependent and bilateral, they are found in about 10 percent of patients
younger than 6 years of age, 50 percent of patients younger than age 30, and almost all
patients by age 50. Other ocular findings in neurofibromatosis include optic nerve gliomas
(the most common CNS tumor) and congenital glaucoma. The frequency of optic gliomas in
neurofibromatosis has ranged from 15 to 20 percent.Patients usually present with symptoms
of decreased visual acuity or visual field defects but may initially appear with signs and
symptoms of increased intracranial pressure. The tumor mass can involve the optic chiasm
or hypothalamus and rarely manifests as the diencephalic syndrome of infancy.Although
there is some controversy regarding the nature of the tumor, optic gliomas in children are
probably congenital hamartomas, indolent and slow-growing.
Congenital glaucoma, a known complication of neurofibromatosis, is commonly associated
with a neurofibroma of the superior eyelid. The mechanisms underlying its cause include
angle obstruction by neurofibromas, angle narrowing from neurofibromatous thickening of
the ciliary body and choroid, fibrovascularization and synechial narrowing of the angle,
and developmental anomalies of the angle.
A variety of bony changes can occur in neurofibromatosis, including a ballooning of the
middle fossa, an enlarged sella turcica, a J-shaped sella, and abnormalities of the
sphenoid wing. The optic foramina are enlarged in some patients with optic glioma; bony
defects of the orbit, the area of the lambdoid sutures, and other cranial bones are not
uncommon. Scoliosis is reported in 10 to 40 percent of patients, although usually it is
not reported before the age of 6 years; kyphosis, anterior meningocele, enlarged
intervertebral foramina, bowing of the tibia and fibula, and bony overgrowth have also
been reported. Pseudarthrosis is a characteristic feature of neurofibromatosis and usually
involves the distal tibia, although other tubular bones can be affected.The bony defects
associated with neurofibromatosis are secondary to developmental abnormalities of
mesenchymal tissue and are usually not the result of bone erosion by tumor.
Magnetic resonance imaging (MRI) of the head may show focal areas of increased signal
intensity in T2-weighted images that are not associated with vasogenic edema and are seen
primarily in the basal ganglia, internal capsule, brainstem, cerebellum, and subcortical
white matter. They tend to involute spontaneously with age.
Other features of neurofibromatosis include macrocephaly and short stature, which are
reported to occur in 10 to 40 percent of patients. Mental retardation and seizures, though
not necessarily related, occur in about 10 percent of patients, and 40 percent of patients
have specific learning disabilities and hyperactivity.Precocious puberty has been observed
in patients with a hypothalamic glioma or hamartoma or in some optic chiasmal gliomas that
involve the hypothalamus. It should not be presumed, however, that precocious puberty
without hypothalamic involvement by mass lesion is an integral part of the disease.
Hypertension can develop as a result of intimal proliferation and fibromuscular changes of
the media in small renal arteries.
A variety of tumors occur more frequently than in the general population, including
pheochromocytoma, leukemia, neuroblastoma, and Wilm's tumor. There is an increased
frequency of multiple endocrine neoplasia and medullary thyroid carcinoma. Among patients
with pheochromocytomas, 4 to 23 percent have neurofibromatosis, whereas fewer than 1
percent of patients with neurofibromatosis have been found to have pheochromocytomas.
The frequency of neurofibromatosis, inherited as an autosomal dominant trait, is about 1
in 4,000 persons for type 1 and about 1 in 50,000 persons for type 2. About one-half of
the cases are said to be spontaneous mutations. The factors behind the risk of developing
the varied complications of the disease are yet to be determined. The gene for type 1 has
been mapped to a locus on chromosome 17; for type 2 it has been localized to chromosome
22. The mutation of the neurofibromatosis type 1 gene on chromosome 17 results in the gene
product neurofibromin, which is partially homologous to guanosine triphosphatase
activating protein. Mutations of the neurofibromatosis type 2 gene on chromosome 22 result
in a protein product schwannomin. The neurofibromatosis type 2 gene suppresses tumor
formation, which accounts for CNS tumors in patients with type 2 neurofibromatosis.
The treatment of patients with neurofibromatosis is symptomatic. Peripheral neurofibromas
are generally indolent lesions that do not require surgical removal unless they are
subjected to repeated trauma or show rapid growth. On occasion, plexiform neuromas are
removed for cosmetic reasons. Intracranial and intraspinal tumors are treated with
appropriate surgical techniques, irradiation, or chemotherapy. Optic gliomas in children
are generally thought to be hamartomas; some authors recommend that they be managed
conservatively by following their growth and documenting visual function, rather than by
immediate surgery or irradiation therapy
|