 |
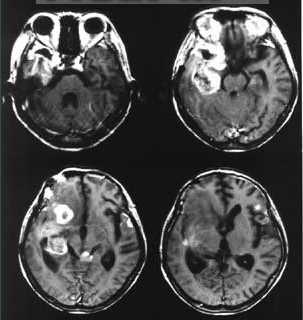
Gliosarcoma. Etiology: It is thought that the sarcomatous changes may arise from fibroblasts of the blood vessels. Gross: The glioblastoma part of the tumor forms heterogeneous infiltrative areas with hemorrhage and necrosis. The sarcomatous portion produces a firm and more discrete mass. Microscopic: Gliosarcoma is a variant of glioblastoma multiforme with a biphasic pattern. There are alternating areas of glial and mesenchyma differentiation. The glial portions show the typical features of a GBM. The sarcomatous areas can resemble either a fibrosarcoma with bundles of spindle cells in a herringbone pattern or a malignant fibous histiocytoma with giant cells. Other mesenchyma tissues can be seen including cartilage, bone, smooth and striated muscle. Epithelial differentiation has also been noted. Staining: The sarcomatous portion should demonstrate reticulin. The glial portion should stain for GFAP. Many gliosarcomas stain for factor-VIII related antigen and Ulex europaeus. Incidence: Rare tumor, occurs in approximately 2% of GBMs. |
| According to the new World Health Organization (WHO) classification
gliosarcoma is defined as a glioblastoma variant characterized by a biphasic tissue
pattern with alternating areas displaying glial and mesenchymal differentiation.
Gliosarcoma is a relatively rare malignant neoplasm accounting for about 2% of all the Glioblastomas. They usually affect the adult population in the fourth to the sixth decade of life. Males are more frequently affected than females (M:F ratio 1.8:1). Gliosarcomas are usually located in the cerebral cortex involving the temporal, frontal, parietal, and occipital lobe in decreasing frequency. Invariably the clinical history of the patient is short and the presenting symptoms depend upon the location of the tumor. The etiology of gliosarcoma remains speculative although it is recognized that gliomas can induce sarcomatous transformation in the supporting mesenchymal elements and irradiation of the central nervous system can induce malignant transformation of the brain parenchyma and the meninges predominantly to fibrosarcoma. Grossly Gliosarcoma may be poorly delineated peripheral grayish tumor mass with central yellowish necrosis stippled with red and brown of recent and remote hemorrhage with the sarcomatous component producing firm discrete mass. The diagnosis of gliosarcoma is based on a biphasic tissue pattern composed of two distinct malignant cell populations, one component being gliomatous (fulfilling the criteria for glioblastoma) and the other with malignant mesenchymal differentiation (fulfilling the criteria for being a sarcoma). Interestingly the sarcomatous component can have varied histological features ranging from a herring bone pattern of fibrosacroma to that of malignant osteiod and cartilaginous differentiation of an osteosarcoma and chondrosarcoma respectively. In the above case the sarcomatous component of the tumor showed features of fibrosarcoma with further mesenchymal differentiation to osteogenic sarcoma and chondrosarcoma. Another diagnostic criterion to be fulfilled before a tumor is classified as Gliosarcoma is the demonstration of GFAP through immunohistochemistry in the gliomatous portion and reticulin in the sarcomatous component preferably exhibiting a clear demarcation between the sarcomatous and the glial component. Additional immunohistochemical stains like smooth muscle actin, desmin, cytokeratin etc can be performed to further identify the mesenchymal differentiation. Although one component of gliosarcoma is clearly of astrocytic origin the histogenesis of the sarcomatous component is still controversial. Feign et al in 1955 described gliosarcoma as glioblastoma in which the proliferating tumor vessels acquired features of a sarcoma. This view was widely accepted, given the prominent vascular proliferation found in glioblastoma. Several immunohistochemical studies supported this hypothesis by demonstrating the presence of factor VIII related antigen and Ulex europaeus I agglutinin in the sarcomatous component while other studies failed to confirm these findings. Another hypothesis suggested a process of dedifferentiation within the glioma with secondary loss of GFAP _expression and acquisition of mesenchymal characteristics. Another school of thought regards gliosarcoma as a spindle cell or desmoplastic variant of glioblastoma (4). However these views have not been supported by recent genetic analysis, which point to a monoclonal origin where p53 mutation, PTEN mutations, p16 (CDKN2A) deletions and coamplification of MDM2 and CDK4 have been identified both in the gliomatous and the sarcomatous component. Gliosarcoma has the same prognosis has Glioblastoma and should be treated identically. |
| Cancer. 1995 Jun 15;75(12):2910-8. |
Clinicopathologic features of primary and postirradiation cerebral gliosarcoma.
Perry JR, Ang LC, Bilbao JM, Muller PJ.
Division of Neurology, Sunnybrook Health Sciences Centre, Toronto, Ontario, Canada.
BACKGROUND. Gliosarcoma is an uncommon malignant brain tumor with mixed glial and mesenchymal elements. Experience is limited to case series, and pathologic data are disparate, leading to uncertainty about clinical features, management, and histogenesis. METHODS. A clinicopathologic review of 32 patients with survival analysis and immunohistochemical studies was performed including glial fibrillary acidic protein analysis, alpha-1-antitrypsin (alpha-1-AT) analysis, and smooth muscle actin (SMA) analysis. RESULTS. Twenty-five patients had primary gliosarcoma, whereas 7 developed gliosarcoma after irradiation for glioblastoma multiforme (GBM). Clinical features were similar to those of GBM. Most tumors were intraaxial and diffusely infiltrating by radiologic studies and at surgery. Median survival for primary gliosarcoma was 25 weeks overall, with patients who received irradiation surviving longer (46 vs. 13 weeks, P < 0.025). Gliosarcoma occurring after irradiation appeared hyperdense by computed tomography in five of seven cases, and median survival was 53 weeks. Primary gliosarcoma was a dimorphic tumor with malignant glial elements and features of malignant fibrous histiocytoma (MFH) or fibrosarcoma and one osteosarcoma. Smooth muscle actin labeled tumor vessels heavily, but in 15/25 primary cases, it extended to the surrounding spindle cells. The remaining cases appeared morphologically like MFH and tended to be positive for alpha-1-AT. Postirradiation gliosarcoma was fibrosarcomatous with positive SMA in 75% of the cases examined. CONCLUSIONS. Gliosarcoma behaves clinically like GBM, and survival may be improved by cranial irradiation of selected patients. Smooth muscle actin reactivity in sarcomatous areas suggests histogenesis in some tumors from the smooth muscle within GBM, whereas others may arise via different mechanisms including differentiation from a pluripotential precursor. Transformation of the smooth muscle within GBM may have therapeutic implications for antiangiogenesis agents.
| Radiother Oncol. 2001 Oct;61(1):57-64. |
Gliosarcoma: a clinical study.
Lutterbach J, Guttenberger R, Pagenstecher A.
Germany.
BACKGROUND AND PURPOSE: Gliosarcomas are rare biphasic neoplasms of the central nervous system composed of a glioblastoma multiforme (GBM) admixed with a sarcomatous component. There are conflicting reports regarding their clinical aggressiveness. Four hundred and twenty-two consecutive patients with GBM were treated at our hospital between 1980 and 1999, among them 12 gliosarcomas. The goal of this study was to examine clinical features, treatment, survival and patterns of failure of gliosarcoma patients and to compare them with the entire group of GBM patients. This comparison was refined by a matched pair analysis with a group of 12 GBM patients selected for age, Karnofsky performance status, resection status, fractionation scheme and total dose (control GBM group). MATERIALS AND METHODS: Seven gliosarcoma patients were male, five female, with a median age of 56 years (range 37-76 years). The median tumor size was 4.5 cm (range 3-8 cm). The locations, all supratentorial, included temporal in six, parietal in five, frontal in four and occipital in one patient. All patients underwent tumor resection followed by postoperative radiation therapy. RESULTS: Median survival was 11.5 months for the gliosarcoma group, 8.1 months for the entire GBM group (log rank test, P=0.16) and 11.0 months for the control GBM group (log rank test, P=0.36). All gliosarcoma patients had local tumor recurrences and died due to neurologic causes within 19.3 months after radiation therapy. CONCLUSIONS: With regard to clinical features, survival and patterns of failure, gliosarcomas and GBM cannot be distinguished clinically. Therefore, the same principles should be applied for the treatment of these tumors.
| J Neurosurg. 1998 Sep;89(3):425-30. |
Clinical outcome of gliosarcoma compared with glioblastoma multiforme: North Central Cancer Treatment Group results.
Galanis E, Buckner JC, Dinapoli RP, Scheithauer BW, Jenkins RB, Wang CH, O'Fallon JR, Farr G Jr.
Department of Oncology, Mayo Clinic and Mayo Foundation, Rochester, Minnesota 55905, USA.
OBJECT: Gliosarcoma, a rare malignancy of the central nervous system, consists of gliomatous and sarcomatous elements. There are conflicting reports regarding its aggressiveness and cell line of origin compared with those of glioblastoma multiforme (GBM). The goal of this study was to compare clinicopathological features such as disease-free survival time and actual survival time in patients with gliosarcoma with a matched group of patients with GBM as well as with the entire group of patients with GBM. METHODS: The authors report on 18 cases of gliosarcoma derived from a series of 748 Grade 4 astrocytoma cases that were part of four consecutive randomized Phase III trials conducted between 1979 and 1996. In this series the gliosarcoma group represented only 2.4% of all GBMs and included 11 men and seven women with a median age of 61.5 years (range 31-81 years). The median tumor size was 5 cm (range 2-8 cm). The locations, all supratentorial, included temporal in 44%, parietal in 28%, frontal in 17%, and occipital in 11%. The 18 patients with gliosarcomas, all Grade 4 (World Health Organization classification), were compared with the entire group of 730 patients with GBM and a control group of 18 patients with GBM matched for known prognostic factors including patient age, randomization date, performance status, extent of resection, and protocol number. Patients in all treatment groups received radiation and nitrosourea-based chemotherapy. The median time to progression and the median survival times for the patients with gliosarcoma were 28.0 and 35.1 weeks as compared with 24.7 and 41.6 weeks for the entire group of patients with GBM (log rank test, p = 0.94 and 0.27, respectively) and 16.7 and 34.4 weeks in the control group (p = 0.20 and 0.84, respectively). In previous molecular cytogenetic analyses of gliosarcoma these authors have shown similar genetic changes in the gliomatous and sarcomatous components. CONCLUSIONS: The data obtained in this study support the conclusion that gliosarcoma shares significant clinical and genetic similarities with GBM and that the same principles should be applied for patient enrollment in research protocols and treatment for these two kinds of tumor.
| Cancer. 1991 May 1;67(9):2342-9. |
Mixed glioblastoma multiforme and sarcoma. A clinicopathologic study of 26 radiation therapy oncology group cases.
Meis JM, Martz KL, Nelson JS.
Department of Soft Tissue Pathology, Armed Forces Institute of Pathology, Washington, DC 20306-6000.
Twenty-six cases are reported of gliosarcoma (GS) retrieved from a series of 1479 glioblastomas (GBM) that were part of five consecutive, randomized Phase II or III malignant glioma protocols initiated by the Radiation Therapy Oncology Group between 1974 and 1983. The clinicopathologic features of these 26 cases, including actuarial survival times, were compared with the remaining 1453 GBM. The minimal qualitative and quantitative histologic criteria required to diagnose GS are presented. In most cases the sarcomatous component was a malignant fibrous histiocytoma; a minority were fibrosarcoma. No significant differences between GS and GBM were found with regard to age, sex, pretreatment Karnofsky performance status, tumor location, size, median survival (8.3 and 9.6 months, respectively), and actuarial survival. None of the treatment regimens, which included various combinations of radiation therapy and chemotherapy, improved the survival of GS over GBM. Selective involvement of the temporal lobe by GS was not found, and the frequency of GS was determined to be only 1.8% of all GBM.
| Br J Neurosurg. 1995 Apr;9(2):171-8. |
Primary cerebral gliosarcoma: report of 17 cases.
Parekh HC, O'Donovan DG, Sharma RR, Keogh AJ.
Department of Neurosurgery, Royal Preston Hospital, UK.
A retrospective study of 17 cases of primary cerebral gliosarcoma is presented. These uncommon highly aggressive intracranial neoplasms were seen at the Royal Preston Hospital, between 1973 and 1992. The patients' ages ranged from 21 to 73 years (mean 52), nine were males and eight were females. They presented with signs and symptoms of a rapidly expanding brain tumour. The diagnosis was suspected on radiological findings and confirmed by histological examination. Treatment involved surgical excision in 15 cases and biopsy in two followed by radiotherapy. Chemotherapy was given in three cases. Despite active management, median survival was only 9 months. The clinical, radiological and pathological features of these lesions are highlighted with emphasis on combined histochemistry and immunohistochemistry. The features of gliosarcoma and glioblastoma are compared and contrasted.