|
|
||
|
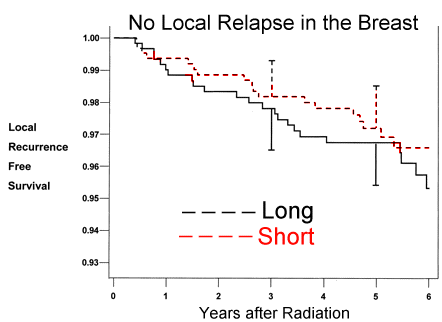
Results: From April 1993 through September 1996, 1234 women were randomly assigned to treatment, 622 to the short arm and 612 to the long arm. Median follow-up was 69 months. Five-year local recurrence-free survival was 97.2% in the short arm and 96.8% in the long arm (absolute difference = 0.4%, 95% confidence interval [CI] = –1.5% to 2.4%). No difference in disease-free or overall survival rates was detected between study arms. The percentage of patients with an excellent or good global cosmetic outcome at 3 years was 76.8% in the short arm and 77.0% in the long arm; the corresponding data at 5 years were 76.8% and 77.4%, respectively (absolute difference = –0.6%, 95% CI = –6.5% to 5.5%). Conclusion: The more convenient 22-day fractionation schedule appears to be an acceptable alternative to the 35-day schedule.
Study Patients
Women with invasive carcinoma of the breast treated by lumpectomy with pathologically negative axillary lymph nodes were eligible for the trial. Patients were excluded from the study for the following reasons: 1) level I and II axillary dissection not performed; 2) presence of invasive or intraductal carcinoma involving the inked margin of excision on pathologic examination; 3) presence of a tumor of more than 5 cm in diameter or clinical T4 disease; 4) presence of multicentric disease; 5) previous diagnosis of breast cancer; 6) presence of bilateral malignancy of the breast; 7) breast deemed too large to permit satisfactory radiation therapy (i.e., the maximum width of breast tissue >25 cm); 8) patient currently pregnant or lactating; 9) presence of serious nonmalignant disease (e.g., cardiovascular or pulmonary) that would preclude radiation treatment; 10) diagnosis of previous or concomitant malignancies of any type except squamous or basal cell carcinoma of the skin and carcinoma in situ of the cervix; 11) patient geographically inaccessible for follow-up; 12) presence of psychiatric or addictive disorders that would preclude informed consent or adherence to the protocol; 13) patient not treated with chemotherapy who was unable to commence radiation therapy within 16 weeks of the last surgical procedure on the breast; 14) patient treated with chemotherapy who was unable to commence radiation therapy within 8 weeks of the last dose of chemotherapy; and 15) patient enrolled in another clinical trial.
Consecutive eligible patients presenting at participating centers who met the inclusion criteria were registered. Participating centers included the Cancer Care Ontario Regional Cancer Centres in the cities of Hamilton, Toronto, Ottawa, Sudbury, London, Windsor, Kingston, and Thunder Bay; the Princess Margaret Hospital in Toronto; and the Montreal General Hospital. Reasons for noneligibility were documented. Written informed consent was obtained from eligible patients before assignment to treatment. The study protocol was approved by the institutional review board of each participating center.
Treatment Regimens
Patients were assigned to one of two
regimens, according to a prescribed
computer-generated central randomization schedule
within strata defined by age (<50 years or
![]() 50 years), tumor
size (
50 years), tumor
size ( 2 cm or
>2 cm), adjuvant systemic therapy (tamoxifen, any
chemotherapy, or no therapy), and center. Before the
randomization procedure, patients were assessed
for adjuvant systemic therapy, according to the
guidelines of each center. Suggested guidelines
for premenopausal patients stated that chemotherapy should
be considered if two of the following three tumor
characteristics were present: tumor size of more
than 2 cm, poorly differentiated tumor, or
estrogen receptor-negative status. The guidelines
for postmenopausal women stated that adjuvant tamoxifen
therapy should be considered if the tumor was
greater than 1 cm and had an estrogen
receptor-positive status. Patients who received
adjuvant chemotherapy completed chemotherapy before
radiation therapy.
2 cm or
>2 cm), adjuvant systemic therapy (tamoxifen, any
chemotherapy, or no therapy), and center. Before the
randomization procedure, patients were assessed
for adjuvant systemic therapy, according to the
guidelines of each center. Suggested guidelines
for premenopausal patients stated that chemotherapy should
be considered if two of the following three tumor
characteristics were present: tumor size of more
than 2 cm, poorly differentiated tumor, or
estrogen receptor-negative status. The guidelines
for postmenopausal women stated that adjuvant tamoxifen
therapy should be considered if the tumor was
greater than 1 cm and had an estrogen
receptor-positive status. Patients who received
adjuvant chemotherapy completed chemotherapy before
radiation therapy.
Patients were randomly assigned to receive whole breast irradiation of 42.5 Gy in 16 fractions over 22 days or to receive whole breast irradiation of 50 Gy in 25 fractions over 35 days. Radiation therapy was delivered daily, from Monday through Friday. The intention was to treat the breast at risk and the underlying chest wall. Patients were treated in the supine position with the ipsilateral arm raised above the shoulder and immobilized. The treatment volume was irradiated by two opposed tangential fields. The medial border was located at the midsternal line. The lateral border was at the midaxillary line to include the breast with a 1- to 2-cm margin and to limit the amount of lung at the central plane to less than 3 cm. The superior border was located at a horizontal line drawn through the supersternal notch, and the inferior border was located at a horizontal line 1–2 cm below the inframammary fold. Wedge compensation was used to ensure a uniform dose distribution throughout the target volume. A contour was taken at the central plane, and a dose distribution was obtained. The treatment volume was treated uniformly to a given dose plus or minus 7%. The dose was prescribed at a point midway along the central plane, two thirds of the distance from the skin to the base of tangent fields. Portal films were obtained in the treatment position with a therapeutic beam to confirm adequate coverage. Patients were treated with a 4- to 6-megavolt linear accelerator or with cobalt-60 radiation. In this trial, no attempt was made to treat the axilla or the supraclavicular or internal mammary lymph nodes, and boost radiation was not used.